Practice Essentials
Epidermolysis bullosa (EB) is a group of inherited bullous disorders characterized by blister formation in response to mechanical trauma. Historically, epidermolysis bullosa subtypes have been classified according to skin morphology. [1, 2]
Discoveries of the molecular basis of epidermolysis bullosa have resulted in the development of diagnostic tools, including prenatal and preimplantation testing. Based on a better understanding of the basement membrane zone (BMZ) and the genes responsible for its components, newer treatments (eg, gene or protein therapy) may provide solutions to the skin fragility found in patients with epidermolysis bullosa.
Related articles include Epidermolysis Bullosa Acquisita and Pediatric Epidermolysis Bullosa.
Signs and symptoms
Important general points include age of onset; size, frequency, and location of blisters; possible inciting factors; prior diagnostic attempts; prior therapies; and extent of pain or pruritus.
Review of systems information that can be associated with different epidermolysis bullosa (EB) subtypes includes alteration of growth or development and evidence of mucosal involvement, including oral (which is demonstrated in the image below), nasopharyngeal, ocular, genitourinary, GI, or respiratory symptoms. A family history of blistering disease is an important finding to identify.
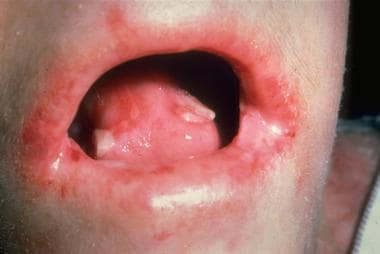 Recessively inherited dystrophic epidermolysis bullosa; oral cavity blistering and scarring.
Recessively inherited dystrophic epidermolysis bullosa; oral cavity blistering and scarring.
Perform a complete physical examination with an emphasis on inspection of all skin, as well as conjunctival, oral, and genital mucosae. Evaluate the size, location, and character of blisters. Attempt to assess the general level at which lesions split. Usually, superficial blisters manifest as crusted erosions, intraepidermal blisters are flaccid and may expand under pressure, and intralamina lucida blisters are tense and heal with atrophy but no scarring. Sublamina densa blisters heal with scarring and milia formation. Assess for involvement of nails, hair, or teeth.
For full information on signs and symptoms of EB subtypes, please see Physical Examination.
Diagnostics
Also see Procedures.
Obtain a skin biopsy following a thorough history and physical examination. Routine histologic analysis is useful only for excluding other causes of blistering. When epidermolysis bullosa (EB) is suspected, the best approach is to obtain 2 biopsy specimens. Analyze one specimen using electron microscopy (EM) and the other using immunofluorescent microscopy.
Evaluate anemia using CBC count with iron studies in patients with severe epidermolysis bullosa, especially recessively inherited epidermolysis bullosa.
Evaluate infection using bacterial cultures from poorly healing wounds or wounds that appear infected.
Imaging studies
Evaluate GI dysfunction. Esophageal strictures associated with junctional epidermolysis bullosa, dystrophic epidermolysis bullosa, or the pyloric atresia associated with a rare form of junctional epidermolysis bullosa can be visualized best by an upper GI series or endoscopy.
Other testing
Evaluate nutrition using serum albumin, height and weight curves, diet diaries, and other analyses of nutrition and growth in patients with severe epidermolysis bullosa.
Evaluate contractures by establishing the range of motion of limbs and digits to monitor contractures and effectiveness of physical therapy.
Routine light microscopy can be used only to exclude other causes of blistering and cannot be used to make the diagnosis of EB.
Management
For a full discussion, please see Treatment.
In May 2023, the US Food and Drug Administration (FDA) approved Vyjuvek (beremagene geperpavec), a topical gene therapy drug, for wound treatment in dystrophic epidermolysis bullosa with COL7A1 mutations. Prolonged use of steroids is contraindicated in the treatment of inherited forms of EB. Steroid-induced complications further warrant prohibiting their use. No other drugs, including phenytoin and tetracycline, have improved the blistering or epithelial disadhesion in EB significantly or consistently.
A topical gel, Filsuvez (birch triterpenes), was approved by the FDA in December 2023 for treatment of wounds in patients aged 6 months and older with junctional and dystrophic epidermolysis bullosa.
Pathophysiology
Epidermolysis bullosa is a family of bullous disorders caused by an absence of basement membrane components due to underlying gene mutations. Epidermolysis bullosa is classified into four major categories: (1) epidermolysis bullosa simplex (intraepidermal skin separation), (2) junctional epidermolysis bullosa (skin separation in lamina lucida or central BMZ), (3) dystrophic epidermolysis bullosa (sublamina densa BMZ separation, as in the images below), and (4) Kindler syndrome (extremely rare, blistering at any level). [3, 4]
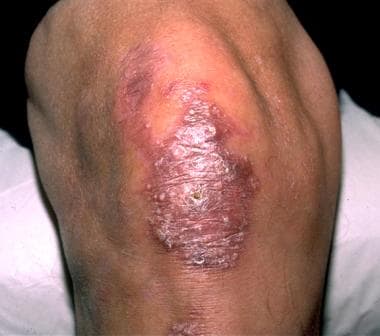 Dominantly inherited dystrophic epidermolysis bullosa. The blistering in this disease often is localized and is characterized by scarring and milia in healed blister sites.
Dominantly inherited dystrophic epidermolysis bullosa. The blistering in this disease often is localized and is characterized by scarring and milia in healed blister sites.
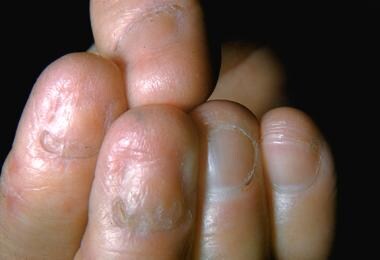 Dominantly inherited dystrophic epidermolysis bullosa. This subtype, similar to other dystrophic and junctional epidermolysis bullosa subtypes, can result in nail dystrophy and loss.
Dominantly inherited dystrophic epidermolysis bullosa. This subtype, similar to other dystrophic and junctional epidermolysis bullosa subtypes, can result in nail dystrophy and loss.
Etiology
Many stratified squamous epithelial tissues, such as the skin and oral mucosa, contain a complex basement membrane zone (BMZ). The BMZ is composed of many specialized components that combine to form anchoring complexes. At the superior aspect of the BMZ, keratin-containing intermediate filaments of the basal cell cytoskeleton insert on basal cell plasma membrane condensations termed hemidesmosomes. Anchoring filaments extend from the basal cell plasma membrane into the extracellular environment and span the lamina lucida, connecting hemidesmosomes with the lamina densa. At the most inferior aspect of the BMZ, type VII collagen‒containing anchoring fibrils extend from the lamina densa into the papillary dermis, connecting the lamina densa to anchoring plaques, trapping interstitial collagen fibrils. Thus, the cutaneous BMZ connects the extensive basal cell cytoskeletal network with the abundant network of interstitial collagen fibrils in the dermis.
Keratin filaments
Keratins 5 and 14 combine to form intermediate filaments in basal keratinocytes. Keratins contain a central alpha-helical rod with several nonhelical interruptions, as well as nonhelical carboxyterminal and aminoterminal regions. The regions of highest conservation between the keratins are located on the ends of the keratin rod in the helix boundary motifs. Keratin intermediate filaments insert upon electron-dense structures termed hemidesmosomes.
Hemidesmosomes
These structures contain intracellular proteins, including plectin and BP230. Plectin (HD1) is a 500-kd protein that binds intermediate filaments. BP230, also termed BPAG1, is a 230-kd protein that has homology to both desmoplakin and plectin. BP230, like plectin, functions in the connection between hemidesmosomes and intermediate filaments. Hemidesmosomes also contain the intracellular portions of the transmembrane proteins collagen XVII (BP180) and alpha-6-beta-4 integrin. The beta-4 integrin subunit performs a central role in hemidesmosome formation and contains an especially large cytoplasmic domain, which interacts with other proteins of the hemidesmosomal plaque. Collagen XVII is a transmembrane collagenous protein that interacts with alpha-4 integrin and BP230 intracellularly and with laminin-332 extracellularly.
Anchoring filaments
These structures contain the extracellular portions of collagen XVII (BP180) and alpha-6-beta-4 integrin. In addition, anchoring filaments contain the molecules laminin-332 and laminin-6. Similar to all members of the family of laminin proteins, laminin-332 is a large heterotrimeric molecule containing alpha-3, beta-3, and gamma-2 chains. Laminin-332 forms a disulfide-bonded attachment to laminin-311, the other known anchoring filament laminin, which contains alpha-3, beta-1, and gamma-1 chains. Laminin-332 also forms a strong association with type VII collagen, which serves to connect anchoring filaments with anchoring fibrils.
Anchoring fibrils
Type VII collagen is the primary component of anchoring fibrils. Type VII collagen contains a large N-terminal globular domain (NC-1), which interacts with laminin-332 in the lamina densa; a long collagenous domain; and a smaller C-terminal globular domain (NC-2), which is cleaved proteolytically during anchoring fibril formation. Type VII collagen chains form a triple helix; then, two molecules join together in an antiparallel fashion. Next, anchoring fibrils are formed by lateral associations of antiparallel dimers. Anchoring fibrils wind around the dermal interstitial collagen fibrils and reinsert back upon the lamina densa, attaching the BMZ to the underlying dermis.
Molecular pathology of epidermolysis bullosa simplex
Most cases of epidermolysis bullosa simplex are associated with mutations of the genes coding for keratins 5 and 14. [5] The level of skin separation is at the mid basal cell associated with variable intermediate filament clumping.
Most epidermolysis bullosa simplex keratin gene mutations are inherited dominantly and interfere with keratin filament assembly. A smaller subset of patients with recessively inherited disease of varying severity exists.
Mutations coding for the most conserved regions of keratins 5 and 14 (helix boundary domains) produce the most severe forms of epidermolysis bullosa simplex. Of the severe forms, the Dowling-Meara subtype exhibits intermediate filament clumping. Conversely, milder forms of the disease, such as the Weber-Cockayne subtype, are associated with mutations at the less conserved regions of keratin 5 and keratin 14 genes. [6]
In patients with epidermolysis bullosa simplex, the mutations that code for the amino terminus of keratin 5 are associated with mottled pigmentation. A small group of patients with recessively inherited epidermolysis bullosa simplex has been shown to have associated muscular dystrophy caused by mutations of the gene coding for HD1/plectin.
Two rare variants of superficial epidermolysis bullosa simplex subtypes have been described, termed lethal acantholytic epidermolysis bullosa simplex (caused by BPAG1/BP230 gene mutations) and ectodermal dysplasia skin fragility syndrome (caused by plakophilin gene mutations). The former is characterized by complete alopecia, severe nail involvement, oral and respiratory tract involvement, and superficial sheetlike separations of the skin. The latter is characterized by generalized erosions, palmoplantar keratoderma, painful fissures, and some nail dystrophy and hair loss. [7]
Molecular pathology of junctional epidermolysis bullosa
Junctional epidermolysis bullosa has a highly variable molecular etiology and represents a collection of different diseases. These diseases all cause blistering in the lamina lucida and variable hemidesmosomal abnormalities. Mutations in genes coding for laminin-332 subunits (alpha-3 chain, laminin beta-3 chain, laminin gamma-2 chain), collagen XVII (BP180), a6 integrin, and b4 integrin have been demonstrated. [6]
More than half of junctional epidermolysis bullosa cases are caused by one of two recurrent nonsense mutations in the LAMB3 gene, which is helpful for mutation analysis and prenatal testing.
Herlitz (letalis) junctional epidermolysis bullosa is characterized by null mutations of laminin-332 genes, resulting in a lack of laminin-332 expression in the tissues of patients.
Missense mutations of laminin-332 genes that result in expression of presumably dysfunctional laminin-332 can result in a milder phenotype, such as generalized atrophic benign epidermolysis bullosa. Generalized atrophic benign epidermolysis bullosa also can be caused by mutations of the gene coding for collagen XVII (BP180).
Mutations of the genes coding for beta-4 and alpha-6 integrin also have been associated with junctional epidermolysis bullosa. In this group of diseases, separation of the skin occurs at the level of the hemidesmosome region. The resultant molecular defects contribute to the clinical manifestation of pyloric atresia.
Molecular pathology of dystrophic epidermolysis bullosa
Dystrophic epidermolysis bullosa thus far has been associated in all cases with mutations of the gene coding for type VII collagen (COL7A1). Anchoring fibrils are affected in patients with dystrophic epidermolysis bullosa, and the degree of involvement ranges from subtle changes to complete absence.
In all patients, a sublamina lucida plane of blister cleavage is present. In some patients, defects of type VII collagen secretion are present.
In the recessive forms, COL7A1 mutations usually cause premature termination codons, resulting in an absence of type VII collagen in tissue. COL7A1 mutations, which do not cause premature termination codons, usually produce less severe disease. For example, mutations that produce glycine substitutions of the triple helical region can interfere with triple helical assembly of the type VII collagen molecule. These types of mutations, which exert a dominant-negative type of effect, are present in many patients with milder dominant forms of this disease. [6]
Molecular pathology of Kindler syndrome
Duplication of the basement membrane is characteristically seen by electron microscopy, along with a variable level of skin separation, either sublamina densa (most common) intralamina lucida, or intraepidermal. Kindlin-1, a protein similar to talin, [8] shows deficient expression in this disease. Mutations of the KIND1 gene coding for kindlin-1 have been associated with this disease. [9, 10, 11]
Epidemiology
Frequency
United States
The United States National Epidermolysis Bullosa Registry [12] found the overall incidence and prevalence of epidermolysis bullosa to be 19.6 and 11.07 cases per 1 million live births, respectively. The incidence and prevalence of epidermolysis bullosa simplex were found to be 7.87 and 6 cases per 1 million live births, respectively. The incidence and prevalence of junctional epidermolysis bullosa were found to be 2.68 and 0.49 cases per 1 million live births, respectively. The incidence and prevalence of dominant dystrophic epidermolysis bullosa were found to be 2.12 and 1.49 cases per 1 million live births, respectively. The incidence and prevalence of recessive dystrophic epidermolysis bullosa were found to be 3.05 and 1.35 cases per 1 million live births, respectively.
International
According to the National Epidermolysis Bullosa Registry, [13] the prevalence of epidermolysis bullosa cases in Norway is 54 cases per million live births, in Japan is 7.8 cases per million live births, in Italy is 15.4 cases per million live births, in Australia is 10.3 cases per million live births, and in Croatia is 9.6 cases per million live births.
Age
Onset of epidermolysis bullosa is at birth or shortly after. The exception occurs in mild cases of epidermolysis bullosa simplex, which may remain undetected until adulthood or occasionally remain undiagnosed.
Prognosis
Epidermolysis bullosa is a lifelong disease. Some subtypes, especially the milder epidermolysis bullosa forms, improve with age.
Infancy is an especially difficult time for epidermolysis bullosa patients. Generalized blistering caused by any subtype may be complicated by infection, sepsis, and death. Severe forms of epidermolysis bullosa increase the mortality risk during infancy. Patients with the generalized severe (previously termed Herlitz or letalis) form of junctional epidermolysis bullosa have the highest risk during infancy, with an estimated mortality rate of 87% during the first year of life. In patients with epidermolysis bullosa who survive childhood, the most common cause of death is metastatic squamous cell carcinoma (SCC), as in the image below.
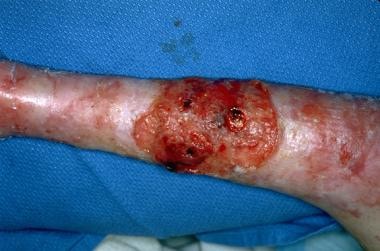
This skin cancer occurs specifically in patients with recessively inherited epidermolysis bullosa who most commonly are aged 15-35 years. In contrast, dominantly inherited epidermolysis bullosa simplex and dystrophic epidermolysis bullosa and milder forms of junctional epidermolysis bullosa may not affect a patient's life expectancy adversely.
One study reported that from 1979-2002, the overall age-adjusted annual mortality annual mortality rate from bullous skin diseases 0.103 death per 100,000 population (2000 US standard population). [14]
Patient Education
Education in proper nutrition and wound care is essential for the patient and family.
-
Epidermolysis bullosa simplex localized (formerly termed Weber-Cockayne subtype). This mild bullous disease is characterized by localized blistering at sites of trauma such as the feet.
-
Epidermolysis bullosa simplex, generalized (formerly termed Koebner subtype). Palmoplantar blistering and hyperkeratosis are noted.
-
Epidermolysis bullosa simplex, generalized (formerly termed Koebner subtype). Close-up image shows hyperkeratotic papules and plaques on the palm.
-
Junctional epidermolysis bullosa, generalized severe (formerly termed Herlitz or letalis) subtype. This severe disease is characterized by generalized intralamina lucida blistering at birth, significant internal involvement, and a poor prognosis.
-
Dominantly inherited dystrophic epidermolysis bullosa. The blistering in this disease often is localized and is characterized by scarring and milia in healed blister sites.
-
Dominantly inherited dystrophic epidermolysis bullosa. This subtype, similar to other dystrophic and junctional epidermolysis bullosa subtypes, can result in nail dystrophy and loss.
-
Recessively inherited dystrophic epidermolysis bullosa pseudosyndactyly (mitten-hand deformity) of the hands and feet.
-
Recessively inherited dystrophic epidermolysis bullosa; oral cavity blistering and scarring.
-
Recessively inherited dystrophic epidermolysis bullosa; squamous cell carcinoma.
-
Diagram illustrating the organization of the dermal epidermal basement membrane and level of disruption in epidermolysis bullosa subtypes. EBS: epidermolysis bullosa simplex. JEB: junctional epidermolysis bullosa. DEB: dystrophic epidermolysis bullosa.









